

近十年来,真性红细胞增多症(polycythemiavera,PV)的诊治认识有了长足的进步。为给我国血液科医师提供规范化的临床实践指导,由中华医学会血液学分会白血病淋巴瘤学组牵头,广泛征求国内专家意见,结合我国现况,反复多次修改,从PV的诊断程序、实验室检查、诊断标准和治疗原则等方面最终达成本共识。
一,诊断程序
1. 病史采集:必须仔细询问患者年龄,有无血管栓塞病史,有无心血管高危因素(如高血压、高血脂、糖尿病、吸烟和充血性心力衰竭),有无疲劳、早饱感、腹部不适、皮肤瘙痒和骨痛,以及活动力、注意力、此前1年内体重下降情况,有无不能解释的发热或重度盗汗及其持续时间,家族有无类似患者,有无长期高原生活史等。建议在患者初诊时及治疗过程中评估疗效时采用骨髓增殖性肿瘤总症状评估量表(MPN-10) 进行症状负荷评估。
2. 实验室检查:以下实验室检查为疑诊 PV 患者的必检项目:
①外周血细胞计数;
②骨髓穿刺涂片和外周血涂片分类计数;
③骨髓活检切片病理细胞学分析和网状纤维(嗜银)染色;
④血清红细胞生成素(EPO)水平测定;
⑤JAK2 V617F和JAK2 第12外显子基因突变检测。有家族病史者建议筛查EPOR、VHL、EGLN1/PHD2、EPAS1/HIF2α、HGBB、HGBA 和 BPGM 等基因突变;
⑥肝脏、脾脏超声或CT 检查。有条件单位可行骨髓细胞体外 BFU-E(± EPO)和CFU-E(± EPO)培养确认是否有内源性红系集落形成。
二,诊断标准
1. PV诊断标准:建议采用WHO(2008)标准。
主要标准:
①男性 HGB>185 g/L,女性 HGB>165 g/L,或其他红细胞容积增高的证据[HGB或红细胞比容(HCT)大于按年龄、性别和居住地海拔高度测定方法特异参考范围百分度的第99位,或如果血红蛋白比在无缺铁情况下的基础值肯定且持续增高至少 20 g/L的前提下男性 HGB>170 g/L,女性HGB>150 g/L];
②有JAK2 V617F突变或其他功能相似的突变(如JAK2第12外显子突变)。
次要标准:
①骨髓活检:按患者年龄来说为高度增生,以红系、粒系和巨核细胞增生为主;
②血清EPO水平低于正常参考值水平;③骨髓细胞体外培养有内源性红系集落形成。
符合2条主要标准和1条次要标准或第1条主要标准和2条次要标准则可诊断PV。
最近,在 WHO(2008)诊断标准的基础上提出的2014年修订建议标准如下:主要标准:
①男性HGB>165 g/L、女性>160 g/L,或男性HCT>49%、女性>48%;
②骨髓活检示三系高度增生伴多形性巨核细胞;
③有JAK2突变。次要标准:血清EPO水平低于正常参考值水平。PV诊断需符合3条主要标准或第1、2条主要标准和次要标准。
2. 真性红细胞增多症后骨髓纤维化(post-PV MF)诊断标准:采用骨髓纤维化研究和治疗国际工作组(IWG-MRT)标准。
主要标准(以下2条均需满足):
①此前按WHO诊断标准确诊为PV;
②骨髓活检示纤维组织分级为2/3级(按0~3级标准)或3/4级(按0~4级标准)。
次要标准(至少符合其中2条):
①贫血或不需持续静脉放血(在未进行降细胞治疗情况下)或降细胞治疗来控制红细胞增多;
②外周血出现幼稚粒细胞、幼稚红细胞;
③进行性脾脏肿大(此前有脾脏肿大者超过左肋缘下5 cm或新出现可触及的脾脏肿大);
④以下 3 项体质性症状中至少出现 1 项:过去6个月内体重下降>10%,盗汗,不能解释的发热(>37.5 ℃)。
三,预后判断标准
PV患者确诊后,为了更好地指导治疗选择,应对患者的预后分组作出判断。采用Tefferi等提出的预后分组积分系统:依年龄(≥67岁为5分,57~66岁为 2 分)、WBC>15 × 109 /L(1 分)和静脉血栓 (1分)分为低危组(0分)、中危组(1或2分)和高危组(≥3分)。
四,治疗
1. 治疗目标:PV的治疗目标是避免初发或复发的血栓形成、控制疾病相关症状、预防 post-PV MF和(或)急性白血病转化。多血症期治疗目标是控制HCT<45%。
2. 一线治疗选择
(1)对症处理:静脉放血和骨髓抑制药物对皮肤瘙痒常无效。由于热水洗澡可使之加重,应告诫患者减少洗澡次数或避免用过热的水洗澡。阿司匹林和塞庚定有一定疗效,但抗组胺药物无效。
(2)血栓预防:由于栓塞是PV患者的主要死亡原因,因此,确诊患者均应进行血栓预防。首选口服低剂量阿司匹林(100 mg/d),不能耐受的患者可选用口服潘生丁。
(3)静脉放血:一般来说,开始阶段每 2~4 d 静脉放血400~500 ml,HCT降至正常或稍高于正常值后延长放血间隔时间,维持红细胞数正常(HCT<45%)。HCT>64%的患者初期放血间隔期应更短,体重低于50 kg的患者每次放血量应减少,合并心血管疾患的患者应采用少量多次放血的原则。静脉放血可使头痛等症状得到改善,但不能降低血小板和白细胞数,对皮肤瘙痒和痛风等症状亦无效。年龄<50岁且无栓塞病史患者可首选此种治疗方法。红细胞单采术可在短时间内快速降低HCT,在必要时可以采用此治疗。反复静脉放血治疗可出现铁缺乏的相关症状和体征,但一般不进行补铁治疗。
(4)降细胞治疗:高危患者应接受降细胞治疗。对静脉放血不能耐受或需频繁放血、有症状或进行性脾脏肿大、有严重的疾病相关症状、PLT>1500×109 /L 以及进行性白细胞增高亦为降细胞治疗指征。
羟基脲或α干扰素(IFN-α)为任何年龄PV患者降细胞治疗的一线药物。在年轻患者(<40 岁)中,羟基脲应慎用。年长患者(>70岁)可考虑间断口服白消安。
羟基脲起始剂量为30 mg·kg-1·d-1,口服1周后改为 5~20 mg·kg-1·d-1,需维持给药并调整用药剂量,联合静脉放血治疗(必要时采用红细胞单采术)可降低栓塞并发症。
IFN-α用药量为(9~25)×106 U/周(分3次皮下注射)。用药6~12个月后,70%患者的HCT可获控制,20%的患者可获部分缓解,10%无效。此外,还可使血小板计数、皮肤瘙痒和脾脏肿大得到显著改善。
3. 二线治疗选择:约25%的患者对羟基脲耐药或不耐受(表1),20%~30%的患者对干扰素不耐受,这些患者可采用二线治疗。
(1)32P:静脉给予32P 2~4 mCi治疗1次常可使疾病得到很好的控制,间隔6~8周后可依首剂疗效再次给予。32P治疗最大的不良反应是远期发生治疗相关性白血病或骨髓增生异常综合征(MDS)及肿瘤。32P治疗后10年的白血病/MDS风险率为10%,肿瘤风险率为15%。20年时白血病或MDS发生风险率可增高至30%。
(2)白消安:2~4 mg/d,口服,几周后常可同时使血小板和白细胞计数下降至正常,停药后血细胞计数维持正常几个月至几年不等。一个大系列研究显示白消安治疗患者的中位首次缓解期为4年。白消安可致严重骨髓抑制,用量不宜超过4 mg/d。
(3)芦可替尼:在一项国际、随机、开放标签、多中心Ⅲ期临床试验中,依赖静脉放血治疗伴有脾脏肿大的 PV 患者随机接受芦可替尼(110 例,起始剂量20 mg/d)或标准治疗(112例,医师根据情况选用羟基脲、干扰素、阿拉格雷、来那度胺、沙利度胺或不予任何治疗),32周时芦可替尼和标准治疗组患者的HCT控制率(HCT<45%)分别为60%和20%,脾脏容积减少 35%的比例分别为 38%和1%,完全血液学缓解率分别为24%和9%,症状下降50%的患者比例分别为49%和5%。据此结果,2014年12月芦可替尼被FDA批准用于治疗羟基脲疗效不佳或不耐受的PV患者。推荐起始剂量为20 mg/d,在开始治疗的前4周不进行剂量调整,每次剂量调整间隔不应少于2周,最大剂量不超过50 mg/d。芦可替尼最常见的血液学不良反应为3/4级的贫血、血小板减少以及中性粒细胞减少,但极少导致治疗中断。治疗过程中外周血PLT<50×109 /L或中性粒细胞绝对值<0.5×109 /L、HGB<80 g/L应停药。停药应在 7~10 d 内逐渐减停,应避免突然停药,停药过程中推荐加用泼尼松(20~30 mg/d)。
4. post-PV MF 和白血病变患者的治疗:post-PV MF的治疗按原发性骨髓纤维化治疗原则,具体参考“原发性骨髓纤维化诊断和治疗中国专家共识(2015版)”。白血病变患者参照相应指南处理。
五,疗效判断标准
根据欧洲白血病网和骨髓增殖性肿瘤研究和治疗国际工作组2013年修订的PV疗效评价标准(表2),主要包括临床血液学及骨髓组织学评价两方面。分子生物学疗效对于评价完全缓解(CR)或部分缓解(PR)不是必需的。完全分子生物学缓解(CRm)定义为:原先存在的异常完全消失。部分分子生物学缓解仅用于基线的等位基因突变负荷≥20%且等位基因突变负荷下降≥50%的患者。
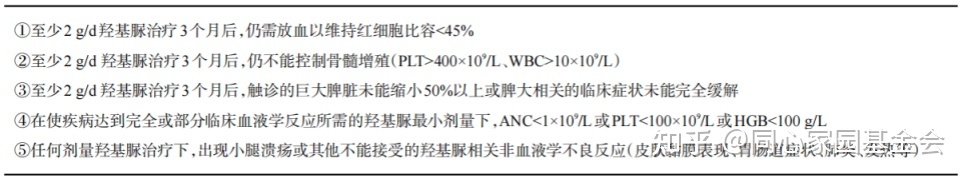
表1 真性红细胞增多症患者羟基脲治疗耐药或不耐受的判断标准

表2 真性红细胞增多症的疗效标准
中华医学会血液学分会白血病淋巴瘤学组
执笔:肖志坚
如有MPN相关疾病患者想交流了解相关疾病知识,请关注MPN家园(志愿者手机及微信号码:18526254316),MPN家园是骨髓增殖性肿瘤病友互动、沟通、学习、互助的平台。MPN家园以患者为中心,开展慈善活动为宗旨,通过为骨髓增殖性肿瘤患者提供专业医疗救助服务,资助相关医学领域学术研究和学术交流,推动MPN疾病治疗的标准化、规范化、便利化,从而使更多病友从中获益。
 微信扫一扫
微信扫一扫 